Step 9
Import the prebuilt perfluoroanthracene |
Start the builder by typing pqsmol at the the command prompt.
As we have already constructed, and optimized, the geometry of perfluoroanthracene, we can start with that. Select the File→Import menu item, Figure ![[*]](TUTORIALS/crossref.png) .
.
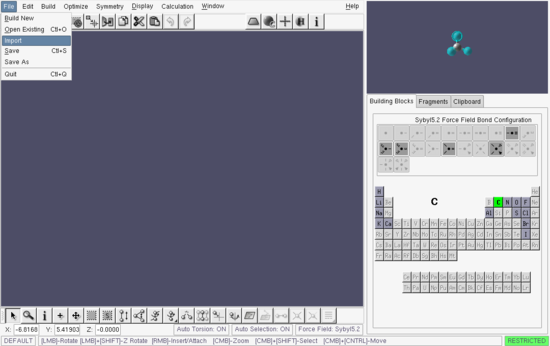
Build window - importing an existing structure into PQSMOL
|
The Import Geometry dialog (Figure ) will open, allowing various file types to be imported. We are going to import the final converged geometry of perfluoroanthracene which is available in the .coord file, F-anthracene.coord. In the File Filter dropdown list select *.coord (coordinates) and select the file F-anthracene.coord from those shown in the Files list.
| Note: As all jobs for this tutorial have been run in advance, the .coord files for all structures are already available; normally of course only the .coord file for perfluoroanthracene - the job run in the first part of this tutorial - would be present.
|
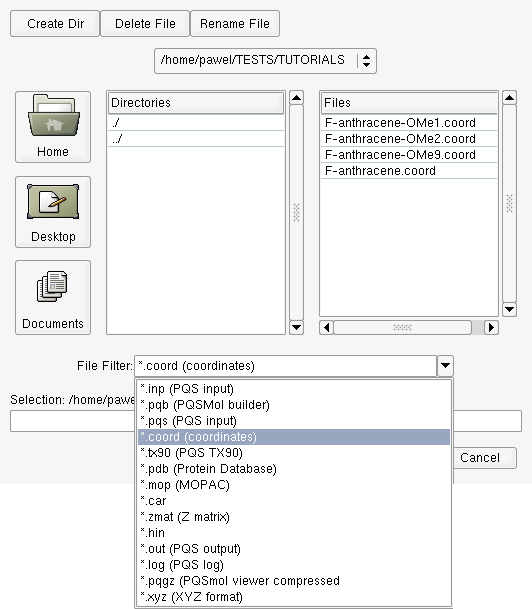
Import dialog - importing a .coord file into PQSMOL
|
As the structure is imported into the Segment buffer the dialog in Figure should appear. When an existing (and compatible) .pqb file is found with the same name as the file being imported, PQSMOL will extract forcefield data (atom types and connectivity) from this file.
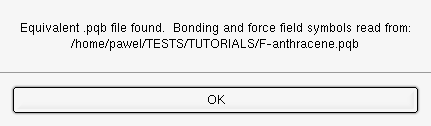
Import info dialog - geometry imported with an existing .pqb file.
|
Note: If a corresponding .pqb is not found in the same directory as the file to be imported, i.e., you are importing raw structural data, then PQSMOL will attempt to derive forcefield data using an internal algorithm. The import info dialog in this case would look like the one in Figure .

Import info dialog - geometry imported without an existing .pqb file.
|
|
Press the OK button to import the structure. The result is given in Figure which shows perfluoroanthracene imported into the Segment buffer. Because of the symmetry of this molecule and the orientation of the coordinate axes in the Segment buffer, the structure is shown "on edge". Drag the mouse pointer in the Segment window to reorient the structure.
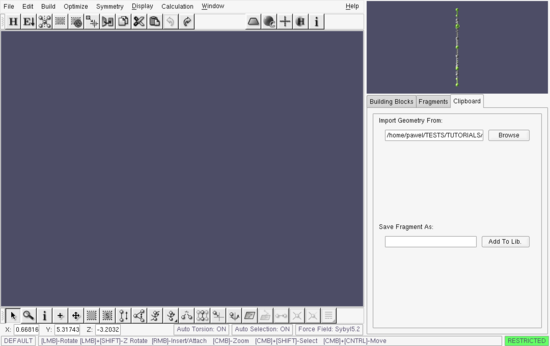
Build window - perfluoroanthracene imported into the Segment buffer
|
Using either the Add/Attach/Connect tool or the overloaded right-click in the Default tool insert the perfluoroanthracene segment into the build area, Figure .

Build window - perfluoroanthracene inserted
|
Step 10
Attach the oxygen building block |
Select the Building Blocks tab in the Segment window and select oxygen in the periodic table. The result is shown in Figure . There are only two possible building blocks for oxygen in restricted mode, either a double bond or a bent configuration with two single bonds, with the latter, which is the one we want, as the default.

Build window - bent oxygen building block selected
|
Using either the Add/Attach/Connect tool or the overloaded right-click in the Default tool attach the oxygen building block to the to top fluorine in the right-most benzene ring. This is the 1-position. The result is shown in Figure .
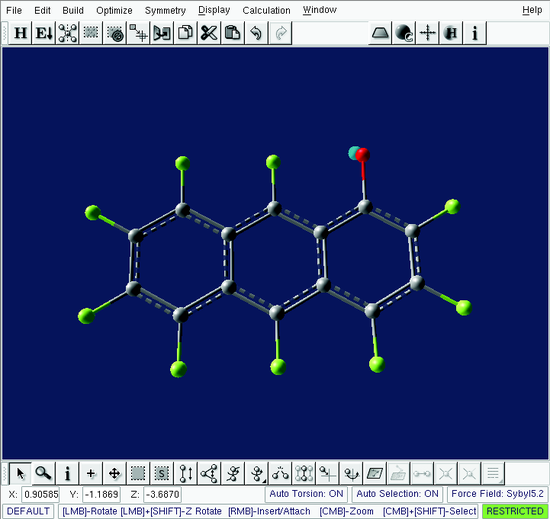
Build window - perfluoroanthracene with the oxygen building block attached at position 1
|
Step 11
Attach the tetrahedral carbon building block. |
In the periodic table select carbon. This will show the five building blocks available for carbon, with the 4-bond sp3 hybridized carbon – again the one we want – the default. Position the mouse pointer over the free valence (the green dummy atom) on the oxygen atom and right click to attach the carbon atom. The result is shown in Figure .

Build window - attaching the tetrahedral carbon building block
|
Finally, press the Hydrogen Fill button  in the top toolbar to fill the three free valencies on the newly attached carbon atom with hydrogens. This completes the building of 1-methoxy-perfluoroanthracene Figure .
in the top toolbar to fill the three free valencies on the newly attached carbon atom with hydrogens. This completes the building of 1-methoxy-perfluoroanthracene Figure .
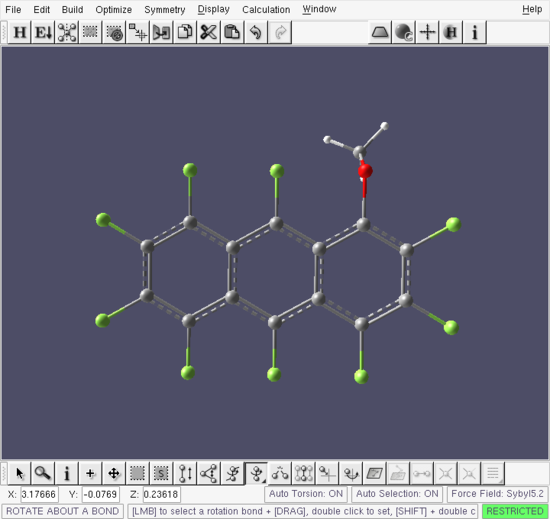
Build window - 1-methoxy-perfluoroanthracene
|
At this point we could optimize the geometry of 1-methoxy-perfluoroanthracene using the Sybyl5.2 force field. However, as we have imported the already-optimized geometry of perfluoroanthracene (optimized at our desired level of theory) and all we have done is replace a fluorine by a methoxy group, then it might be better to start the ab initio optimization using the structure "as is" without the force field preoptimization. (The methoxy group can be rotated about the C-O bond to any desired position.)
We can now repeat Step 6 above and prepare the input file. If desired, you can skip this step and go direct to the examination of the output. The output file is called F-anthracene-OMe1.out.
[PREV] [NEXT]